第一性原理计算在若干铁基合金中的研究进展
崔春娟1,2,赵芷琪1,李昊霖1,乌彤超1,王志聪1(1. 西安建筑科技大学冶金工程学院; 2. 陕西省冶金工程技术研究中心)
摘要:基于密度泛函理论(DFT)的第一性原理计算是研究材料物理化学性质的重要手段,对新材料的发展具有重要意义。这种方法仅需输入元素种类、 原子数和初始结构, 即可预测材料的晶体结构、电子结构和性能。随着计算机技术的进步,中国科学技术大学在“神威·太湖之光”超级计算机上实现万原子分子固体的大规模第一性原理计算。这一突破使得高精度的材料模拟在大尺度、长时间范围内成为可能,为材料研究提供了更精准的预测和模拟。第一性原理计算可帮助理解材料性质、预测不同环境下的材料行为,并指导新材料的发现和设计,有望显著缩短研发周期并降低成本。本文概述了第一性原理计算的理论基础,并详细评述了基于密度函数理论的第一性原理计算方法在若干铁基合金研究中的进展,同时探讨了其中存在的问题和未来发展趋势,为新型铁基合金的计算模拟研究提供了参考。
关键词: 铁基合金; 第一性原理计算; 晶体结构; 力学性能; 磁学性能
目录介绍
1 第一性原理计算的理论基础
1.1 密度泛函理论
1.2 交换关联泛函
1.3 赝势
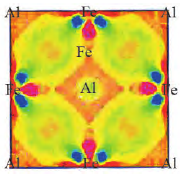
2 第一性原理计算在铁基合金中的应用
2.1 晶体结构
2.2 电子结构
2.3 力学性能
2.4 磁学性能
2.5 表面特性
3 总结与展望
©软件著作权归作者所有。本站所有文件均来源于网络,仅供学习使用,请支持正版!
转载请注明出处!




发表评论 取消回复